


History
A 9-year-old boy attends the paediatric dermatology clinic. Both he and his parents are concerned about an accumulation of facial lesions over several months. His mother has tried several over-the-counter anti-acne remedies without apparent benefit. He is other-wise well, although in comparison with his siblings (two older sisters) he has struggled at school; closer questioning suggests that he has problems academically as well as with his behaviour, although this has never been formally evaluated. He does not take medication. His family are all well, his parents are non-consanguineous.
Examination
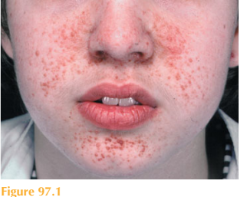
He has symmetrically distributed red to pink papules, each measuring 1–3 mm in diam-eter localized to the nasolabial folds and extending to cheeks, nostrils and also the chin (Fig. 97.1). The individual lesions are smooth and firm. Examination of his skin under Wood’s light demonstrates six hypomelanotic macules over his lower back, abdomen and buttocks. Examination of his cardiorespiratory system reveals a midsystolic ejection murmur. His neurological system is normal including fundoscopy, vision and hearing. He does appear to be easily frustrated and requires significant reassurance from his mother throughout the examination, avoiding eye contact and following only the instructions of his mother. His abdomen is soft and there are no palpable masses. Both of his parents also agreed to full skin examination and no abnormalities were detected.
Questions
• What are these lesions?
• With what are they associated?
• How would you investigate further?
 Medicine
Medicine
These lesions are facial angiofibromas (formerly also called adenoma sebaceum). These lesions, associated with hypomelanotic ‘ash-leaf’ macules, are features of tuberous sclero- sis complex. Other cutaneous features include shagreen patch, fibrous plaques, periungual fibromas and confetti-like hypopigmentation. Autosomal dominantly inherited mutations of two genes, TSC1 (chromosomal locus, 9q34; gene product hamartin) and TSC2 (16p13; tuberin), can cause tuberous sclerosis complex, which has a broad clinical spectrum. There is a high rate of de-novo mutations with 50–70 per cent of cases presenting as such. Promising new systemic treatments known as mTOR inhibitors are being trialled for complications of tuberous sclerosis. Examination of family members, however, can detect features of a less severe phenotype.
Diagnosis is based on a constellation of clinical features. Definite tuberous sclerosis com-plex is diagnosed by the presence of either two major features, or one major feature plus two minor features. Presentation with fewer features may fall into categories of probable or possible tuberous sclerosis. A thorough skin examination is important, as skin signs are identified in the majority of affected individuals. Ash-leaf hypomelanotic macules may be present from birth or infancy. Shagreen patches or facial plaques often appear during the first decade. Facial angiofibromas commonly develop in late childhood or around puberty, often stabilizing at puberty. Periungual fibromas often begin at puberty and may continue to accumulate with age.
The clinical scenario presents four issues that need to be addressed: (1) question mark over neurodevelopment; (2) possible pathological cardiac murmur; (3) potential for other occult hamartomas; and (4) implications for other family members.
1. Further investigation of neurodevelopment would include a multiprofessional assess-ment, ideally within a Child Development Centre, by a community paediatrician, an educational psychologist and a speech therapist. Over 50 per cent of individuals with tuberous sclerosis complex have a low intelligence quotient (IQ) or developmental disorder (particularly autism or language disorders). Unrecognized petit mal seizures can cause problems with school performance, justifying an assessment by a paediatric neurologist with or without electroencephalographic monitoring. A CT scan of the brain would look for evidence of subependymal nodules which may be calcified; MRI is more sensitive in the detection of subependymal giant cell astrocytomas.
2. Further assessment of the cardiac murmur would entail an ECG and echocardiogram to detect cardiac rhabdomyomas. These lesions usually regress with age and are not treated unless of functional significance.
3. Other baseline investigations would include routine blood tests (particularly renal function), renal ultrasound scan, and ophthalmology review (looking for translucent retinal nodules which may represent hamartomas or astrocytomas). Dental pits may also be found.
4. Family members should be offered renal biochemistry and ultrasonography as well as fundoscopy. Even in cases of apparent de-novo mutation unaffected parents of children with tuberous sclerosis complex have a recurrence risk of 1–2 per cent in subsequent pregnancies. Genetic counselling should be initiated.
KEY POINTS
• Tuberous sclerosis complex is one of the most common neurocutaneous disorders.
• The disease has a broad clinical spectrum affecting almost all organ systems.
• It is due to an autosomal dominant mutation in one of two tumour suppressor genes encoding the proteins tuberin and hamartin.
need an explanation for this answer? contact us directly to get an explanation for this answer